|
|
Metazoan complexes |
Emili & Marcotte labs |
- Home
-
H.sapiens
-
M.musculus
-
D.melanogaster
-
C.elegans
-
S.purpuratus
-
Conserved map
-
PPI Projections
- Download
- Help
- Contact
Panorama of ancient metazoan macromolecular complexes
Homo sapiens
Human
Humans (variously Homo sapiens and Homo sapiens sapiens) are primates of the family Hominidae,
and the only extant species of the genus Homo.
Humans are distinguished from other primates by their bipedal locomotion,
and especially by their relatively larger brain with its particularly well developed neocortex, prefrontal cortex and temporal lobes,
which enable high levels of abstract reasoning, language, problem solving, and culture through social learning.
Like all mammals humans are a diploid eukaryotic species.
Each somatic cell has two sets of 23 chromosomes, each set received from one parent, gametes have only one set of chromosomes which is a mixture of the two parental sets. Among the 23 chromosomes there are 22 pairs of autosomes and one pair of sex chromosomes.
Like other mammals, humans have an XY sex-determination system, so that females have the sex chromosomes XX and males have XY. One human genome was sequenced in full in 2003, and currently efforts are being made to achieve a sample of the genetic diversity of the species (see International HapMap Project).
By present estimates, humans have approximately 22,000 genes.
The variation in human DNA is minute compared to that of other species, possibly suggesting a population bottleneck during the Late Pleistocene (ca. 100,000 years ago), in which the human population was reduced to a small number of breeding pairs.
Nucleotide diversity is based on single mutations called single nucleotide polymorphisms (SNPs).
The nucleotide diversity between humans is about 0.1%, which is 1 difference per 1,000 base pairs. A difference of 1 in 1,000 nucleotides between two humans chosen at random amounts to approximately 3 million nucleotide differences since the human genome has about 3 billion nucleotides.
Most of these SNPs are neutral but some (about 3 to 5%) are functional and influence phenotypic differences between humans through alleles
(from Wikipedia).
Mus musculus

House mouse
The house mouse (Mus musculus) is a small rodent, a mouse. It is one of the most numerous species of the genus Mus.
Although a wild animal, the house mouse mainly lives associated with humans, causing damage to crops and stored food.
The house mouse has been domesticated as the pet or fancy mouse, and as the laboratory mouse, which is one of the most important model organisms in biology and medicine.
It is by far the animal most commonly genetically altered for scientific research.
Sequencing of the mouse genome was completed in late 2002.
The haploid genome is about three billion base pairs long (3000 Mb distributed over 20 chromosomes), therefore equal to the size of the human genome.
Estimating the number of genes contained in the mouse genome is difficult, in part because the definition of a gene is still being debated and extended. The current count of primary coding genes is 23,139.
For comparison, humans have an estimated(from Wikipedia).
Drosophila melanogaster

Drosophila melanogaster
Drosophila melanogaster is a species of Fly (the taxonomic order Diptera) in the family Drosophilidae.
The species is known generally as the common fruit fly or vinegar fly. Starting with Charles W. Woodworth's proposal of the use of this species as a model organism, D. melanogaster continues to be widely used for biological research in studies of genetics, physiology, microbial pathogenesis and life history evolution.
It is typically used because it is an animal species that is easy to care for, breeds quickly, and lays many eggs.
Flies belonging to the family Tephritidae are also called fruit flies, which can lead to confusion, especially in Australia and South Africa, where the term fruit fly refers to members of the Tephritidae that are economic pests in fruit production, such as Ceratitis capitata, the Mediterranean fruit fly or "Medfly".
Genome. The genome of D. melanogaster (sequenced in 2000, and curated at the FlyBase database) contains four pairs of chromosomes: an X/Y pair, and three autosomes labeled 2, 3, and 4.
The fourth chromosome is so tiny that it is often ignored, aside from its important eyeless gene.
The D. melanogaster sequenced genome of 139.5 million base pairs has been annotated and contains approximately 15,016 genes.
More than 60% of the genome appears to be functional non-protein-coding DNA involved in gene expression control.
Determination of sex in Drosophila occurs by the X:A ratio of X chromosomes to autosomes, not because of the presence of a Y chromosome as in human sex determination.
Although the Y chromosome is entirely heterochromatic, it contains at least 16 genes, many of which are thought to have male-related functions(from Wikipedia).
Caenorhabditis elegans
An adult Caenorhabditis elegans
Caenorhabditis elegans is a free-living, transparent nematode (roundworm), about 1 mm in length, that lives in temperate soil environments.
Research into the molecular and developmental biology of C. elegans was begun in 1974 by Sydney Brenner and it has since been used extensively as a model organism.
Genome C. elegans was the first multicellular organism to have its genome completely sequenced. The sequence was published in 1998, although a number of small gaps were present; the last gap was finished by October 2002. The C. elegans genome sequence is approximately 100 million base pairs long. The genome consists of six chromosomes (named I, II, III, IV, V and X) and a mitochondrial genome. Its gene density is about 1 gene/5kb.
Introns, or non-expressed sequences, are 26% of the genome. Some large intergenic regions contain repetitive DNA sequences. Many genes are arranged in operons: polycistronic series that are transcribed together. C. elegans and other nematodes are among the few eukaryotes currently known to have operons.
The genome contains approximately 20,470 protein-coding genes. The number of known RNA genes in the genome has increased greatly due to the 2006 discovery of a new class of 21U-RNA genes, and the genome is now believed to contain more than 16,000 RNA genes, up from as few as 1,300 in 2005. Scientific curators continue to appraise the set of known genes, such that new gene predictions continue to be added and incorrect ones modified or removed.
In 2003, the genome sequence of the related nematode C. briggsae was also determined, allowing researchers to study the comparative genomics of these two organisms. Work is now ongoing to determine the genome sequences of more nematodes from the same genus, such as C. remanei, C. japonica and C. brenneri. These newer genome sequences are being determined using the whole genome shotgun technique, which means they are likely to be less complete and less accurate than that of C. elegans, which was sequenced using the "hierarchical" or clone-by-clone approach.
The official version of the C. elegans genome sequence continues to change as new evidence reveals errors in the original sequencing; DNA sequencing is not an error-free process.
(from Wikipedia).
Strongylocentrotus purpuratus

Strongylocentrotus purpuratus
The purple sea urchin, Strongylocentrotus purpuratus, lives along the eastern edge of the Pacific Ocean extending from Ensenada, Mexico to British Columbia, Canada. This sea urchin species is deep purple in color and lives in lower intertidal and nearshore subtidal communities. Along with sea otters and abalones, it is a prominent member of the spectacular kelp forest community. It normally grows to a diameter of about 4 inches and may live as long as 70 years. The purple sea urchin plays many roles. Besides its ecological importance, it is also an important fishery along the west coast of the US and it is one of several biomedical research models in cell and developmental biology. Because of its importance to biomedical research, the sea urchin genome was completely sequenced and annotated in 2006. The sea urchin genome is estimated to encode about 23,300 genes. Many of these genes were previously thought to be vertebrate innovations or were only known from groups outside the deuterostomes. Thus the sea urchin genome provides a comparison to our own and those of other deuterostomes, the larger group to which both echinoderms and humans belong. Using the strictest measure, the purple sea urchin and humans share 7,700 genes. Many of these genes are involved in sensing the environment, a fact surprising for an animal lacking a head structure. These urchins were used for food by the indigenous peoples of California. They ate the yellow egg mass raw (from Wikipedia).
Conserved map
Conserved map
In biology, conserved sequences are similar or identical sequences that occur within nucleic acid sequences (such as RNA and DNA sequences), protein sequences, protein structures or polymeric carbohydrates across species (orthologous sequences) or within different molecules produced by the same organism (paralogous sequences). In the case of cross species conservation, this indicates that a particular sequence may have been maintained by evolution despite speciation. The further back up the phylogenetic tree a particular conserved sequence may occur the more highly conserved it is said to be. Since sequence information is normally transmitted from parents to progeny by genes, a conserved sequence implies that there is a conserved gene. It is widely believed that mutation in a "highly conserved" region leads to a non-viable life form, or a form that is eliminated through natural selection. Highly conserved proteins are often required for basic cellular function, stability or reproduction. Conservation of protein sequences is indicated by the presence of identical amino acid residues at analogous parts of proteins. Conservation of protein structures is indicated by the presence of functionally equivalent, though not necessarily identical, amino acid residues and structures between analogous parts of proteins (from Wikipedia).
Projected PPI across 122 eukaryotic species
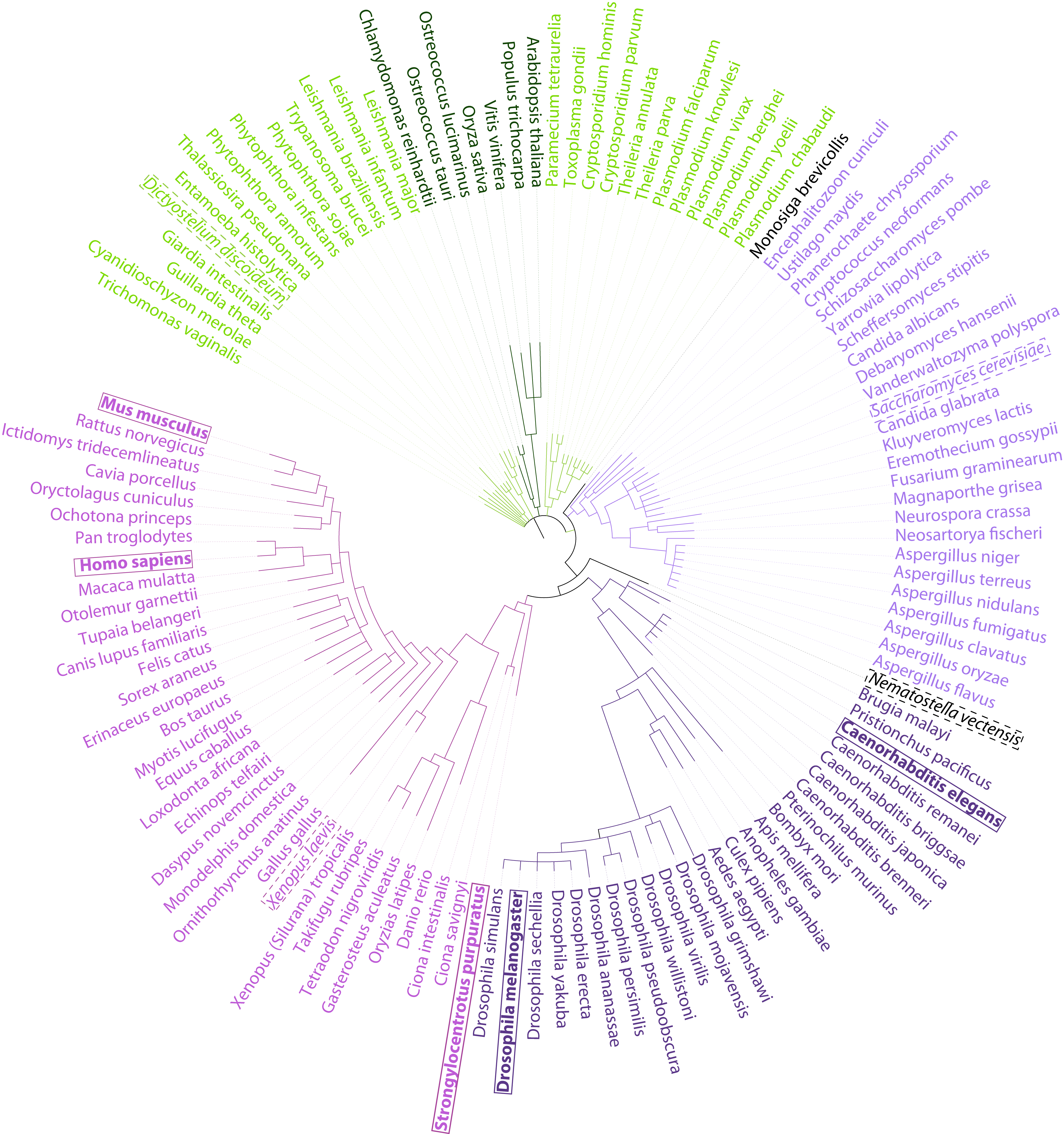
122 sequenced eukaryotic species( Tree generated using phyloT ver. 2014.3 )
Downloads
MS2 Elution Profiles
MS1 Elution Profiles
Orthologs
PPI & Correlations
Predicted & PPI
Complex Network
Help for Metazoan Complexes web-site
Species menu
Conserved map menu
Search: Browse:PPI Projections menu
Search: The web-site is supported by Postgresql database. Manual for the database is here.Search Protein
Protein search allows user to get detailed information about each gene in the list for particular species (Homo sapiens, Mus musculus, Drosophila melanogaster, Caenorhabditis elegans and Strongylocentrotus purpuratus).
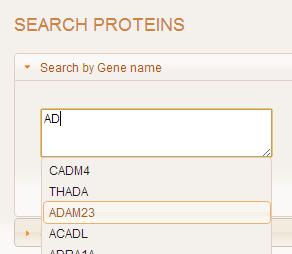
Search by gene name accepts only gene names. If the user uses the auto-complete option to input their query,
then the gene names will be separated by comma. Manually entered gene names can be separated by any character
such as a comma, colon, semi-colon, space, tab, newline or a combination of these options.
If invalid input is entered, an error message is shown and the query is not processed further.
If the input is valid, the gene details page is shown which displays all identifiers, features, and properties.
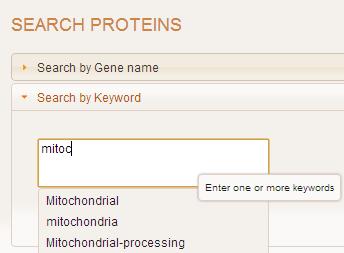
Search by keyword(s) allows users to enter descriptor terms for genes. Examples of acceptable keywords are "Cholinesterase" and "Mitochondrial".
If no input is provided by the user, an error message is shown and is not processed further.
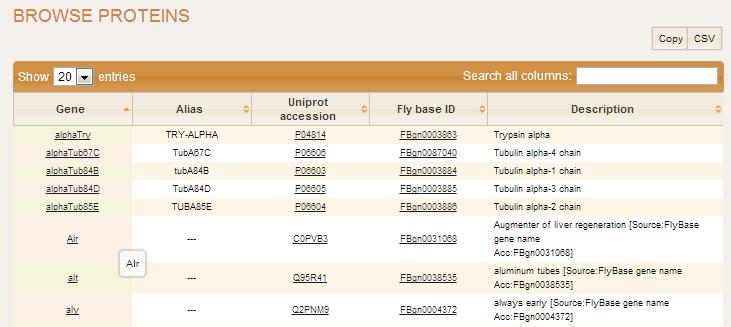
Browse Proteins
Browse Proteins page displays a table consisting of the genes contributing to conserved map in Metazoan Complexes study. It contains gene names, alias, uniprot accession, Ensemble accession or other ID from species-specific database (FlyBase, WormDB, SpBase) along with gene description.
Go to Top
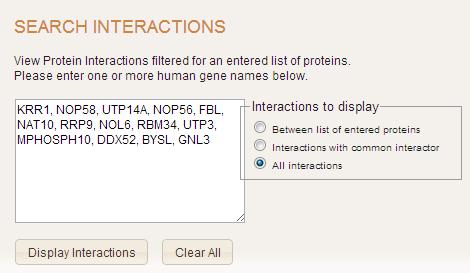
Search Interaction
Search Interactions page allows user to query gene interactions by entering gene names and by choosing interaction type. The gene input area allows to enter multiple gene names to be queried. Manually entered gene names can be separated by any character such as comma, colon, semi-colon, space, tab, newline or a combination of these options. All gene names should be valid names that consists of characters and/or numbers. No other characters are allowed. If this occurs, the user will be prompted with an error.
There are three types of gene interactions:
- All interactions
- Interactions with common interactor
- Interactions between genes
In the first case all possible interactions for genes entered will be displayed. In the second case interactions will be retrieved if there is any gene that interacts with all of the genes from the list. In the last case interactions only within the genes in the list entered will be displayed.
Go to Top
Search Complex
Search complexes page allows user to retrieve detailed information about one or more predicted complexes. Select boxes are available here. User can also type in complex ID in the upper row in the appropriate format (ex. Complex 12) to filter complex IDs. When mouse enters parthicular complex name, information on all complex members in terms of human gene names is displayed in the box on the right.
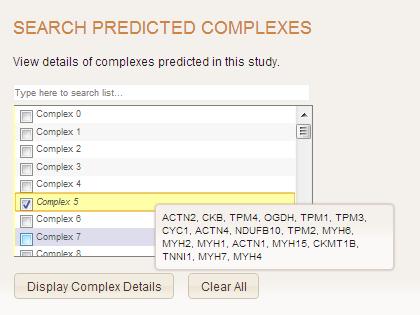
User submits the form by clicking "Display Complex details" button. Then will be directed to a result page that contains a table with detailed information about complex(es) of choice and a link to view complex page, that provides a cytoscape network of the complex. On clicking a plus sign in the first column, nested table with all protein-protein interactions contributing to this complex, will be displayed.
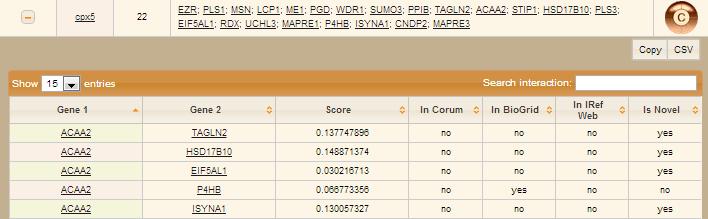
Go to Top
Browse Interactions
Browse interactions page contains a table with entire collection of protein-protein interactions related to Metazoan Complexes study. Detailed information about conserved genes names, score, complexes to which each pair of interacting genes contribute, orthologs in other species and evidence type is provided.
Information can be filtered using any of the search boxes provided below the table columns.
Go to TopBrowse Complexes
This page displays all complexes predicted in this study in one table. Information about complex ID, number of subunits, and members in terms of human gene names is provided. It also contains links to View Complex and View Elution Profile pages. MS1 and MS2 Elution profiles are displayed as an image.
By clicking the plus sign in the first column of the table user can open nested tables displaying complex members in other species and conserved ppi between complex members. Members are mostly presented in terms of gene names for corresponding species. If gene doesn't have name, than its ID is diplayed. In case there is no ortholog gene in this species, dashes are shown. Multiple orthologs are listed in parenthesis with blue font.
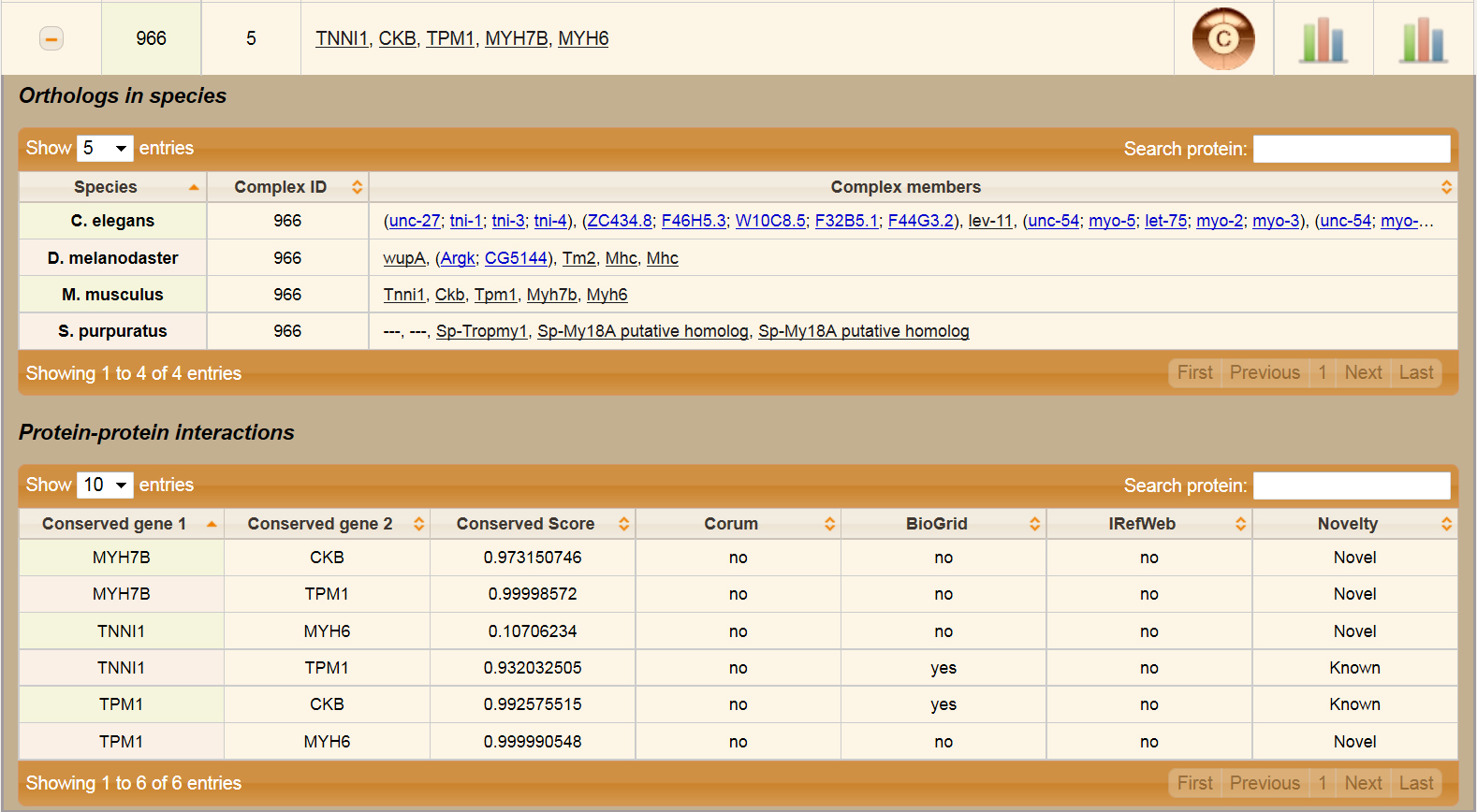
Go to Top
Browse Network page displays all complexes predicted in this study using Cytoscape Web Tool.
Go to TopGene Details page
Gene Details page displays all identifiers, description, orthologs, and information about relevant protein-protein interactioins for particular gene. In IDs section gene identifiers are displayed as links to corresponding public databases (Uniprot, Ensemble, NCBI Gene, etc.).
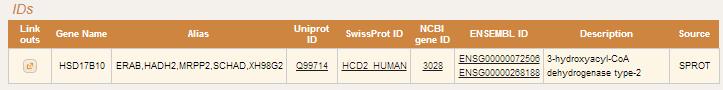
For human genes that have any disease asssociated Disease section will be displayed as a table with two columns - syndrom description and OMIM phenotype ID.

In Orthologs section othologs in other species available for the gene are displayed as link to corresponding gene details page. If there are several orthologs, they all are displayed. If there is no ortholog, dashes are shown.

In interactions contributing to conserved map section relevant interactions are shown as a table. Conserved gene 1 and Conserved gene 2 columns show conserved pair of genes in terms of human gene names. In next two columns orthologs for this pair of genes in corresponding species are displayed. Score, complex IDs to which this interaction contributes, presence in public databases of protein complexes (Corum, IRefWeb, BioGrid) and interaction status (novel or known) are shown.
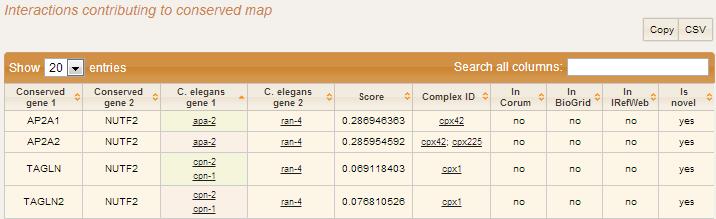
Go to Top
View Complex page
View complex page displays predicted complex using Cytoscape Web Tool and allows user to customize some features.
On clicking a plus sign near complex number Settings panel opens on the left. The panel shows legend and allows user to choose layout (Force Directed, Circle, CoSE, Radial, Tree) and node option to be displayed on Details panel on the right. Both panels are draggable. Details panel is resizable. The following node options are available: gene description, disease, orthologs, phenotypes, essential genes. When option changes to disease, nodes of genes that have any disease associated are highlighted with red color.
Network can be exported and saved on the users computer for future reference in the following formats: PDF, PNG, SVG, XGMML and GraphML.
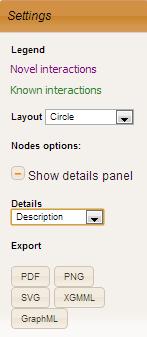
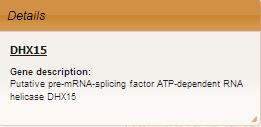
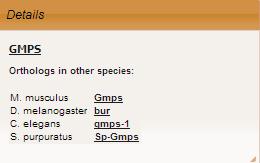
Go to Top
Search PPI projections
Search PPI projections allows the user to select a species from the select list and displays orthology based PPI projections for the ~17K significant ppi from this study.
A table of ppi projections are displayed along with their Human orthologs, the PPI score, complex membership and description. Effort has been made to link the gene/protein/transcript involved in the projected ppi to external reference databases viz. Ensembl, NCBI, Uniprot, JGI portals, EupathDB portals. A download link has been provided to allow the user to download the ppi projections for the species of interest.
Go to Top
Contact Us |
|
E-mail questions and comments to: |
|
Andrew Emili |
Edward M. Marcotte |
E-mail:andrew.emili@utoronto.ca |
E-mail:edward.marcotte@gmail.com |
For help with the website please contact: |
|
Sadhna Phanse |
|
E-mail: sadhna.phanse@utoronto.ca |